Abstract:
Prevalence of Genetic disorders is increasing at an alarming rate worldwide and the issue at hand is to deal with the lack of awareness associated with these disorders. This lack of awareness is the primary reason why early detection and management is not possible today at a large scale.
The objective of this discussion would be to spread information about the disorders and the etiology behind them, and discuss advancements in scientific research which would help in effectively managing the conditions.
Introduction:
The entire human body is designed by genes. It operates through the intricate interaction of the 25,000 genes forming differentiated cells and tissues, carrying out complex reactions, preserving homeostasis and propagating growth.
The stability of human life is maintained through the congruous functioning of every single gene comprising the human genome. Even if a single sequence within any one gene is altered, a huge impact may be seen.
It is because of such importance that today, genetics has become an essential part of modern medicine and treatment.

Figure 1: Defect in gene; image courtesy (Indus Health Plus)
Genetic disorders have become increasingly important in terms of healthcare and quality of living, even more so given the fact that there is an opportunity for early screening, detection and management. Despite this possibility, numerous cases are seen where genetic disorders are either not diagnosed or left untreated.
Thus, a question arises: Where are we Lacking?
The simple answer is, we lack in Awareness.
It is with this purpose in mind that today we discuss two of the common genetic conditions, their complications, pathogenesis, detection and management, including the new developments and strides taken by modern medicine to solve the issue.
Sickle cell anemia
Cystic fibrosis
Main Body:
Sickle Cell Anemia:
Structure of Hemoglobin:
Anemia is a common term which refers to a decrease in the oxygen carrying capacity of blood, but this special type of anemia is caused by a single nucleotide defect in the gene which forms the Hemoglobin molecule.
Hemoglobin (Hb) is a complex structure present inside the Red Blood Cells that transports Oxygen to all tissues. The structure of hemoglobin comprises 4 polypeptide chains (sequence of amino acids). There are two alpha and two beta polypeptide chains in a normal adult Hemoglobin molecule (Hb A). [1]
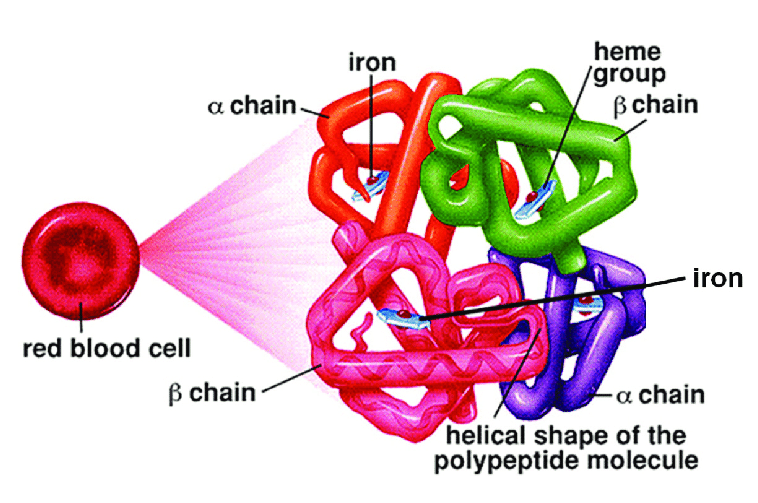
Figure 2: Structure of Hemoglobin molecule; image courtesy (Cetin Gencer, Research Gate)
Mutation:
Now, the interesting details are seen. The entire Hemoglobin molecule is coded by a combination of genes, but one in particular, the HBB gene (Hemoglobin Subunit Beta gene) which is responsible for the formation of the beta subunit [2], has a high chance of getting mutated resulting in an array of genetic disorders such as Thalassemia and Sickle Cell anemia.
In Sickle Cell Anemia, as a result of mutation, Adenine changes to Thymine and as a result the codon changes and Valine is incorporated at the 6th position of the beta polypeptide chains of Hemoglobin in place of Glutamic acid. [2]
This type of Hemoglobin is called Hemoglobin S (Hb S).

Figure 3: Point Mutation in HBB gene resulting in a change in sequence of amino acids in beta polypeptide chain; image courtesy (Understanding Evolution)
This change of amino acids results in a tendency of the hemoglobin molecules to stick to each other at the time of oxygen stress and thus giving the Red Blood Cells their characteristic Sickle shape. Hence the name, Sickle Cell Anemia.[3]
Complications:
The mutated Hemoglobin molecules, when they are not bound to oxygen, stick to each other giving the RBCs a sickle shape. Normally, the red blood cells are discoid shaped and easily pass through the capillaries. These sickle shaped RBCs get stuck in the capillaries leading to occlusion of blood flow. Also, these abnormal RBCs are destroyed inside the spleen and thus have a shorter life span as compared to the 120 days life span of mature RBCs. [3]
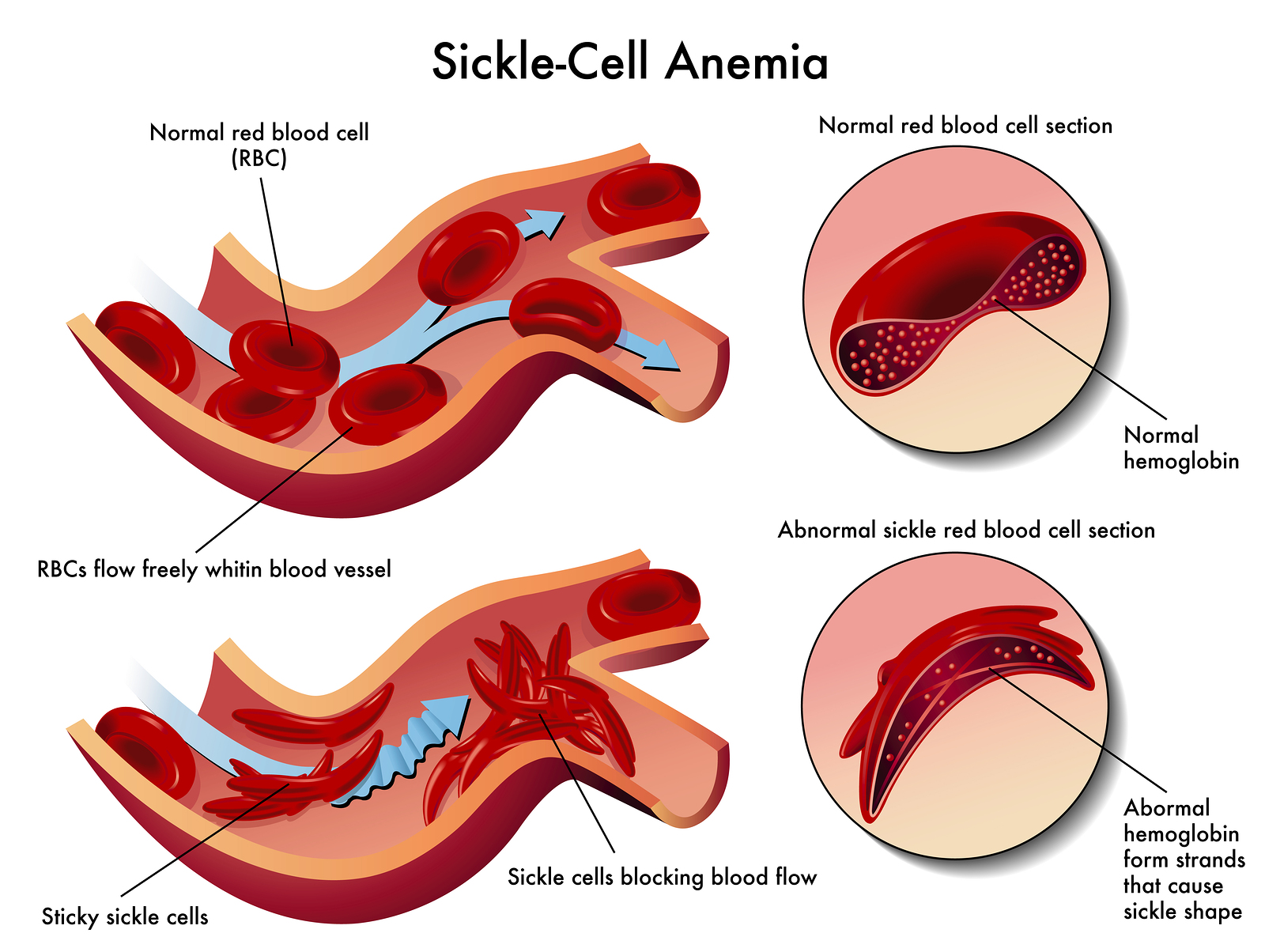
Figure 4: Occlusion of blood vessels due to sickle shaped Red Blood Cells; image courtesy
(Taming The Sru)
Genetics and Inheritance:
As we dive deep into the mutations seen in sickle cell anemia, we see that each individual gets two copies of the HBB gene, one from each parent. These copies are located on the short arm of the 11th chromosome.
The inheritance pattern seen in case of Sickle Cell Anemia is autosomal recessive since the chromosome associated is an autosome and both the defective alleles are required to cause the severe form of the disease.
As a result, two basic conditions can be seen.
The individual gets two defective copies of the HBB gene from both parents. This condition is called Sickle Cell Disease. Such an individual would have predominantly Hemoglobin S in their blood and would start to experience symptoms early in his/her life and can go into crisis. [4]
The individual gets a normal copy of the HBB gene from one parent and a defective copy from the second parent. This condition is called Sickle Cell Trait. Since one normal copy is present both the normal adult Hemoglobin (Hb A) and Hemoglobin S are formed in this individual's blood. Thus, under normal conditions they do not experience any symptoms and might not even be aware of their condition. In extreme situations such as high altitude or severe dehydration, they might develop symptoms of sickle cell anemia.
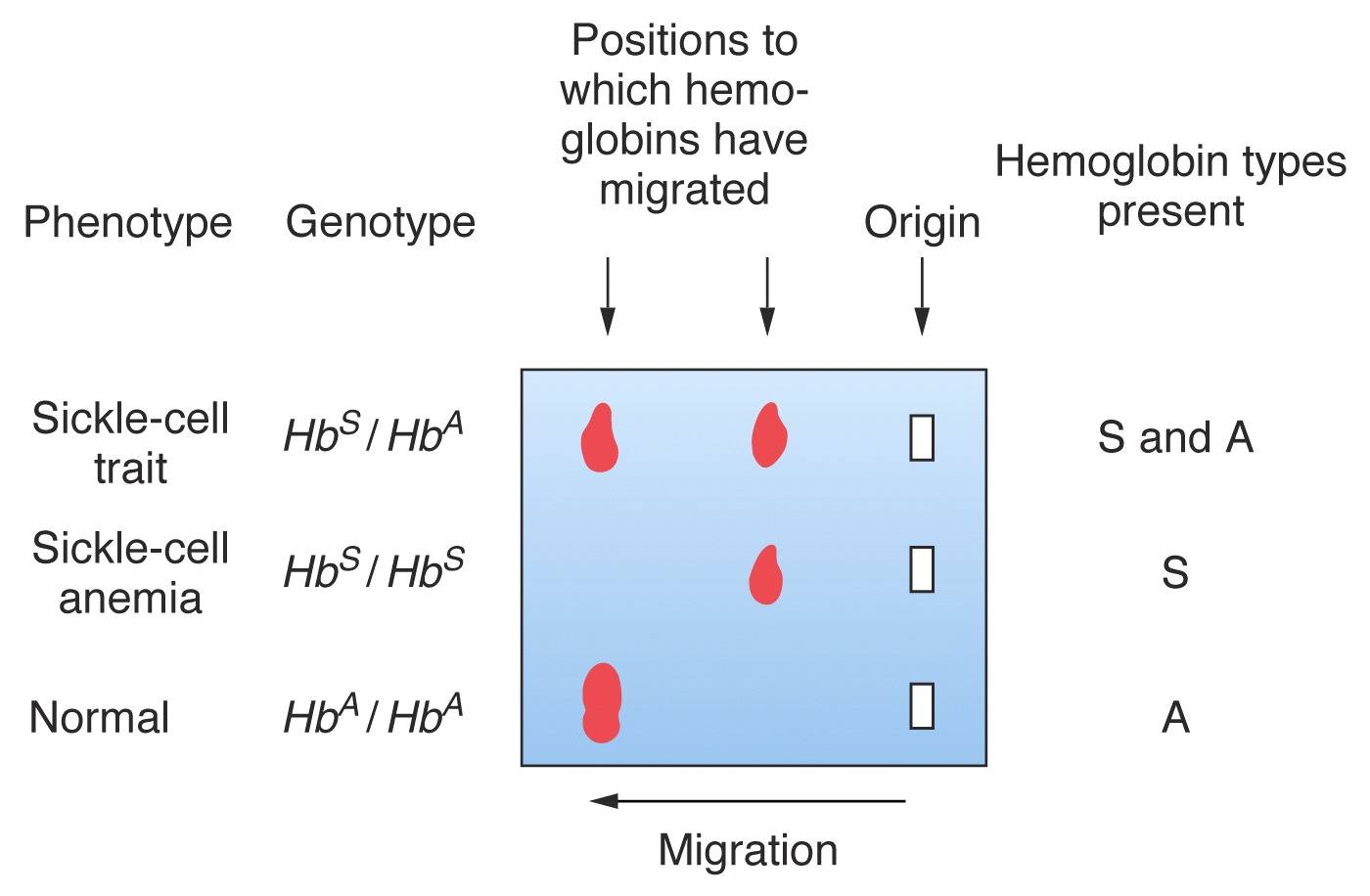
Figure 5: Hemoglobin Electrophoresis results in different genotypes; image courtesy (Steven M. Carr, mun)
Important Associations:
Mutations are not limited and thus, it is seen that Sickle Cell Disease can present in different forms, namely:
HBSS: A condition where both the alleles have the sickle cell mutation
HBSC: Here one allele has the sickle cell mutation and the other allele contains a different mutation. This is a milder form of the Sickle Cell Disease.
HBSbetathalassemia: One allele has the sickle cell mutation and the other allele has the beta thalassemia mutation. [4]
Problem Statement:
Today, Sickle Cell Anemia affects millions, more common in Africa, Middle East and the Indian Subcontinent. It leads to severe complications, poor life quality and increased mortality.
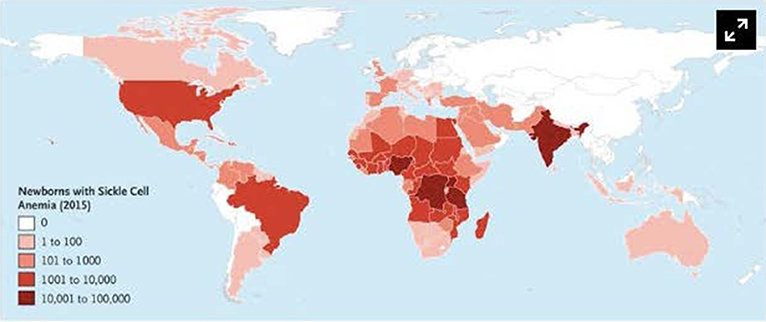
Figure 6: Geographical Distribution of Sickle Cell Anemia; image courtesy (Piel FB, Steinberg MH, Rees DC)
Early Action:
Early detection combined with efficient management has significantly decreased the mortality rates and morbidity associated with Sickle Cell Anemia and further strides are being taken to try and tackle the issue at the genetic level using techniques such as Gene therapy where the formation of fetal Hemoglobin is enhanced using advanced techniques like CRISPR. Fetal Hemoglobin is less prone to sickling and thus helps to maintain the blood flow in the individual's body. [5]
CRISPR is a method of targeting mutated genes and modifying them to look for cures at the genetic level.
Cystic Fibrosis:
Cystic Fibrosis is caused due to a mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. [6]
The CFTR gene codes for the CFTR protein which regulates transport of ions across membranes. A defect in the CFTR gene leads to a defective protein ultimately causing thick mucus to build up over the membranes. [6]
This can lead to severe organ damage including persistent lung infections, difficulty in breathing and failure to thrive.

Figure 7: Building up of thick mucus in Cystic fibrosis; image courtesy (Mayo Foundation)
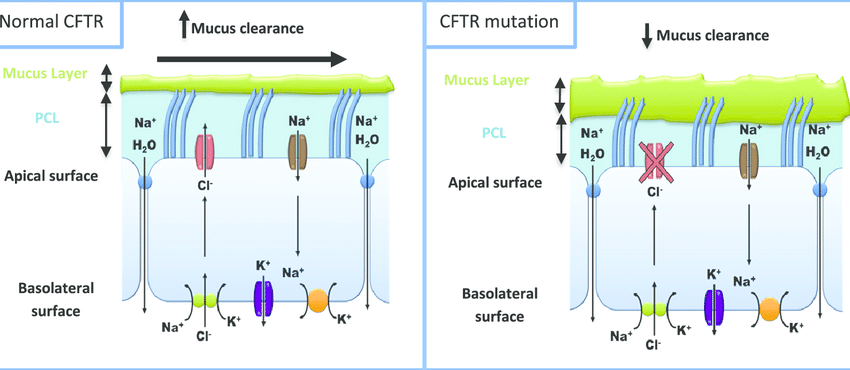
Figure 8: Comparison between normal and mutated CFTR protein; image courtesy (Shankar Kumar, Anand Tana, Anu Shankar)
Mutations:
An enormous complexity is seen in the genetics of Cystic Fibrosis with more than 2000 mutations being identified in the CFTR gene [7]. Mutations in the CFTR gene can be classified into different types based on their mechanism:
Class 1: Protein production mutations
These mutations prevent the formation of the complete CFTR protein. [8]
Class 2: Protein Processing Mutations
Mutations which alter specific sites of the CFTR protein thus stopping its function.
The most noteworthy among these is the F508del mutation which is considered as the most common Cystic Fibrosis mutation. [8]
Class 3: Gating Mutations
These mutations inhibit the transfer of ions through the CFTR protein. [8]
Class 4: Conduction mutations
Mutations which alter the structure of the protein prevent its functioning. [8]
Class 5: Insufficient Protein mutations
Reduces the amount of CFTR proteins on cell surfaces. [8]
The inheritance pattern in Cystic fibrosis is Autosomal Recessive and a defective copy of the gene from both the parents is required to cause the disease.
It is common among the caucasian population. [6]
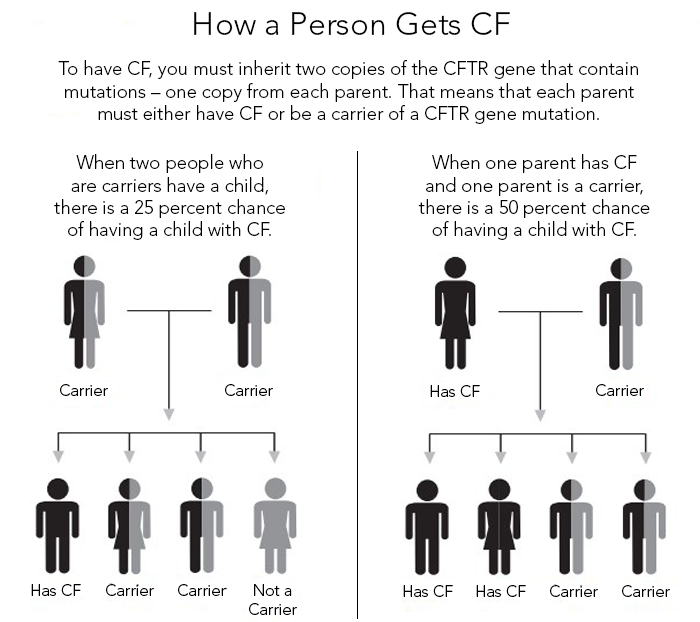
Figure 9: Inheritance Pattern in Cystic fibrosis; image courtesy (Cystic Fibrosis Foundation)
Forward Strides:
New therapies for Cystic fibrosis have been developed which target the CFTR gene defect and have been shown to improve lung function and reduced severity.
Combined with early detection, the majority of the issue can be handled helping human life to prevail.
Conclusion:
The main takeaway from this discussion is that not only are genetic disorders becoming increasingly common but they can also be managed effectively.
The onus lies on the society to spread awareness and promote healthcare facilities such as screening tests, treatment centers and counseling programmes.
Effect of a defective gene is not limited to an individual, it impacts the whole population.
References:
1)Gell D. A. (2018). Structure and function of haemoglobins. Blood cells, molecules & diseases, 70, 13–42. https://doi.org/10.1016/j.bcmd.2017.10.006
2)Mandal, A. K., Mitra, A., & Das, R. (2020). Sickle Cell Hemoglobin. Sub-cellular biochemistry, 94, 297–322. https://doi.org/10.1007/978-3-030-41769-7_12
3)Sundd, P., Gladwin, M. T., & Novelli, E. M. (2019). Pathophysiology of Sickle Cell Disease. Annual review of pathology, 14, 263–292. https://doi.org/10.1146/annurev-pathmechdis-012418-012838
4)Santosh L. Saraf, Robert E. Molokie, Mehdi Nouraie, Craig A. Sable, Lori Luchtman-Jones, Gregory J. Ensing, Andrew D. Campbell, Sohail R. Rana, Xiao M. Niu, Roberto F. Machado, Mark T. Gladwin, Victor R. Gordeuk,
Differences in the clinical and genotypic presentation of sickle cell disease around the world,
Paediatric Respiratory Reviews,
Volume 15, Issue 1,
2014,
Pages 4-12,
ISSN 1526-0542,
https://doi.org/10.1016/j.prrv.2013.11.003.
5)Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y. S., Domm, J., Eustace, B. K., Foell, J., de la Fuente, J., Grupp, S., Handgretinger, R., Ho, T. W., Kattamis, A., Kernytsky, A., Lekstrom-Himes, J., Li, A. M., Locatelli, F., Mapara, M. Y., de Montalembert, M., Rondelli, D., Sharma, A., … Corbacioglu, S. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. The New England journal of medicine, 384(3), 252–260. https://doi.org/10.1056/NEJMoa2031054
6)López-Valdez, J. A., Aguilar-Alonso, L. A., Gándara-Quezada, V., Ruiz-Rico, G. E., Ávila-Soledad, J. M., Reyes, A. A., & Pedroza-Jiménez, F. D. (2021). Cystic fibrosis: current concepts. Fibrosis quística: conceptos actuales. Boletin medico del Hospital Infantil de Mexico, 78(6), 584–596. https://doi.org/10.24875/BMHIM.20000372
7)De Boeck K. (2020). Cystic fibrosis in the year 2020: A disease with a new face. Acta paediatrica (Oslo, Norway : 1992), 109(5), 893–899.
https://doi.org/10.1111/apa.15155.
8)Chaudary N. (2018). Triplet CFTR modulators: future prospects for treatment of cystic fibrosis. Therapeutics and clinical risk management, 14, 2375–2383. https://doi.org/10.2147/TCRM.S147164
Very informative article
ReplyDeleteSuch a beautiful writer
ReplyDelete